quarta-feira, 14 de maio de 2014
MICROCEFALIA
Causas e tipos
.jpg)
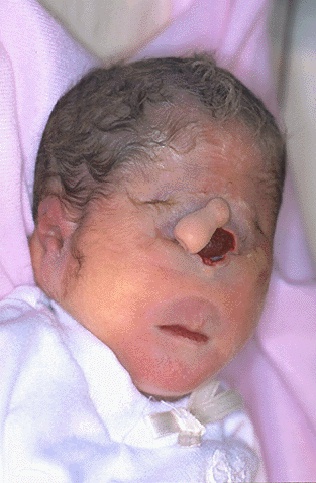
A microcefalia é provocada por uma
insuficiência no desenvolvimento do crânio e do encéfalo, que dá origem a um
crânio de tamanho reduzido e a um cérebro inferior ao normal. Embora possa ser
provocada por várias situações, é possível distinguir dois tipos de
microcefalia.
A microcefalia primária é provocada
por uma anomalia genética. Trata-se de um defeito congénito que, embora seja
muito pouco frequente, é muito grave, pois provoca um escasso desenvolvimento
cerebral, que em alguns casos pode provocar a morte do paciente. Apesar de as
crianças afectadas terem um crânio pequeno, com dimensões semelhantes às do
maxilar inferior, as orelhas e o nariz são proporcionalmente muito grandes.
A microcefalia secundária pode ser
provocada por várias perturbações. Em alguns casos, pode ser originada por um
defeito no desenvolvimento embrionário consequente de várias infecções sofridas
pela mãe ao longo da gravidez (rubéola, toxoplasmose) ou por uma exposição a
radiações ionizantes durante os primeiros meses de gestação. Noutros casos, o
problema é provocado por uma fusão prematura dos ossos do crânio
(craniosinostose), de causa desconhecida, relacionada com outros defeitos
congênitos ou consequente de doenças ósseas como o raquitismo.
.jpg)
As repercussões da microcefalia são distintas,
estando essencialmente dependentes do insuficiente desenvolvimento do cérebro,
não obstante este seja directamente provocado por um defeito no desenvolvimento
embrionário ou consequência da sua impossibilidade em se expandir, co-mo
normalmente ocorre durante a primeira infância.
A microcefalia primária costuma provocar
hipertonia muscular generalizada, paralisia, crises convulsivas e atraso
mental.
As consequências da microcefalia secundária
dependem do tipo e gravidade da malformação. Como as funções cerebrais estão,
na grande maioria dos casos, pouco desenvolvidas, esta situação provoca um
atraso mental profundo. Por vezes, caso apenas seja afectado o desenvolvimento
de um hemisfério cerebral, acaba igualmente por provocar um atraso mental,
apesar de as perturbações motoras serem unilaterais.

Incidência
A microcefalia é uma doença pouco frequente,
pois apenas afecta cerca de 1 em cada 40 000 recém-nascidos.
Fontanelas

Em
condições normais, o crânio, no momento do nascimento, tem um perímetro de
aproximadamente 33 a 36 cm, aumentando de tamanho ao longo dos primeiros anos
de vida, acompanhando o crescimento do encéfalo. Este crescimento é bastante
pronunciado nos primeiros seis meses, entre 7 a 8 cm, até alcançar os 46 a 48
cm no final do primeiro ano. Este aumento de tamanho do crânio apenas é
possível porque os vários ossos que o compõem não estão unidos na altura do
nascimento, algo que apenas acontece posteriormente. Esta situação é evidente,
sobretudo, numa zona da cabeça do bebé denominada fontanelas, que como ainda
não se encontram revestidas pelo osso (como é o caso da fontanela anterior,
situada na parte mais elevada e central do crânio, sendo a última a ser fechada
pouco antes do final do segundo ano de vida) são sensíveis ao tacto. A regular
medição do perímetro craniano e a palpação das fontanelas permitem ao médico
controlar o desenvolvimento do crânio e do encéfalo.
Síndrome
de Edwards ou Trissomia do 18
Descrita
em 1960 por John H. Edwards, hoje a trissomia do 18 apresenta trissomia regular
sem mosaicismo, isto é, cariótipo 47, XX ou XY, +18. Pode haver uma
translocação envolvendo todo ou a maior parte do cromossomo 18, capaz de ser
original ou herdada de um genitor portador balanceado. A trissomia também pode
estar presente na forma de mosaico, com uma expressão variável mas geralmente
mais leve. Ainda não se identificou a “região crítica” da trissomia do 18, mas
a trissomia parcial de todo o braço longo produz o fenótipo típico da trissomia
do 18. A incidência é de cerca de 0,3 por 1000 nascimentos.
A
Trissomia do 18 está associada à idade materna, pois grande parte dos casos são
originados de mulheres com mais de 35 anos de idade.
Características dos Portadores


Os
portadores apresentam retardamento físico e mental, defeitos cardíacos. O
crânio é muito alongado na região occipital. O pescoço é curto. O pavilhão das
orelhas é dismórfico, com poucos sulcos. A boca é pequena e triangular. Grande
distância intermamilar. Os genitais externos são anômalos. O dedo indicador é
maior do que os outros e flexionado sobre o dedo médio. Os pés têm as plantas
arqueadas. As unhas costumam ser hipoplásticas e atrofiadas.
A
morte ocorre em geral antes da primeira infância, aos 3 ou 4 meses de idade,
mas pode ser protelada há quase 2 anos.
Doenças associadas
Como
em todas as trissomias e anomalias genéticas, há sempre doenças associadas e, a
maioria delas são muito graves e de difícil tratamento. A síndrome de Edwards
não é uma excepção, têm várias doenças associadas, entre as quais estão
presentes:
*
Doenças cardíacas;
*
Doenças cerebrais;
*
Deficiência Mental;
*
Patologias Osteo - Auriculares (problemas nas articulações);
*
Mielomeningocelo (é um defeito do desenvolvimento do tubo neural em que existe
herniação da espinhal medula através de um defeito ósseo da coluna vertebral);
*
Anomalias renais (rim em forma de ferradura);
*
Anomalias do aparelho reprodutor.
Como
dá para constatar, são todas doenças complicadas e algumas até raras e
impossíveis de tratar o que torna os portadores desta trissomia mais
vulneráveis à morte.
Cariótipo
.jpg)
Diagnóstico
Pode ser diagnosticada através de ultra-sons
durante a gravidez pode detectar anomalias, porém, isto pode ser confirmado
através da realização de uma amniocintese. As crianças nascidas com
trissomia 18 apresentam, normalmente, um tamanho reduzido ao nascimento. Após o
nascimento, médicos especializados conseguem identificar a doença através da
observação das seguintes características: protuberância na parte posterior da
cabeça, fissuras mais acentuadas na parte exterior dos olhos, boca e maxilar de
dimensões reduzidas, orelhas modificadas, alteração ao nível do tamanho do
punho e dos seus respectivos dedos.
Tratamento
Não há tratamento para a síndrome de Edwards, mas tratamento através de medicamentos para combater alguns dos sintomas são indispensáveis. O tratamento desta doença está direcionado para a proveniência de uma boa nutrição e o tratamento de infecções que são bastantes frequentes e ainda medicamentos que possibilitem um melhor funcionamento do coração do doente. Muitas das crianças nascidas com esta doença apresentam uma grande dificuldade nas questões da alimentação, assim sendo a comida pode ser fornecida através de tubos intra-gástricos ou diretamente através de uma gastrostomia. Para além da parte física do tratamento, é indispensável um bom acompanhamento emocional e psíquico através dos familiares e amigos. As crianças possuem uma esperança de vida muito curta, muitas delas não sobrevivem para além de um ano de vida.
terça-feira, 13 de maio de 2014
Hidrocefalia
O
líquido cefaloraquidiano (líquor) é produzido constantemente dentro dos
ventrículos cerebrais. Em pessoas
normais, o líquor normalmente flui através de vias de um ventrículo ao próximo,
e então para fora do cérebro, descendo para a medula espinhal.
Se
as vias de drenagem do líquor forem obstruídas em algum ponto, o fluído se
acumula nos ventrículos do cérebro, causando neles um inchaço - resultando na
compressão do tecido ao redor. Em bebês e crianças, a cabeça se alargará; em
crianças mais velhas e adultos, o tamanho da cabeça não aumenta porque os ossos
que formam o crânio já estão completamente unidos.
.jpg)

Causas:
A
Hidrocefalia é causada pela inabilidade de drenagem do líquor na corrente
sanguínea. Existem muitas razões pelas quais isto pode acontecer:
Tumor
cerebral - Tumores do cérebro causam inchaço dos tecidos circundantes,
resultando em pobre drenagem do líquor.
Meningite
- Esta é uma infecção das membranas que recobrem o cérebro. A inflamação e
debridação desta infecção pode bloquear as vias de drenagem causando
hidrocefalia.
Hidrocefalia
Congênita- A hidrocelia, neste caso, está presente no nascimento mas isto não
significa que ela seja hereditária.
Prematuridade
- Bebês nascidos prematuramente são mais vulneráveis ao desenvolvimento de
hidrocefalia do que aqueles nascidos a termo, desde que muitas partes do corpo
ainda não estão amadurecidas. A atividade da área que está logo abaixo da
linha dos ventrículos no cérebro apresenta um rico suprimento sanguíneo. Seus
vasos sanguíneos podem ser facilmente rompidos se o bebê sofrer uma mudança na
pressão sanguínea ou na quantidade de fluído no sistema.
.jpg)
Tipos de Hidrocefalia:
Hidrocefalia
Comunicante - Obstrução do fluxo do líquor no espaço subaracnóide após sair do
quarto ventrículo. As causas incluem infecções como meningite (a fibrose
obstrui o espaço subaracnóide e o fluxo do líquor), bem como falha de absorção,
hemorragia subaracnóide ou bloqueio do sangue através de aneurismas.
Hidrocefalia
Não-Comunicante- Obstrução do fluxo do líquor no sistema ventricular ou na
parte externa do foramen. Geralmente, os sítios de estreitamento são
obstruídos. Exemplos incluem cistos colóides os quais obstruem o terceiro
ventrículo e tumores do tronco encefálico os quais comprimem o canal entre o
terceiro ventrículo e o quarto ventrículo (Aqueduto cerebral, ou de Sylvius).
Hidrocefalia
Ex-Vacuo: Algumas vezes o cérebro se retrai em tamanho (como no caso da
Doença de Alzheimer), e, como resultado, os ventrículos se alargarão para
compensar. Ainda que os ventrículos estejam dilatados, eles não estão sob
pressão.
 Tratamento:
Tratamento:
O tratamento
mais comum para a hidrocefalia é a chamada derivação - um tubo plástico
inserido inteiramente dentro da pele que cria uma nova via para o líquor, do
cérebro a outra parte do corpo. A derivação controla a pressão por drenar o
excesso de líquor, prevenindo então, o agravamento da condição.
Entretanto, a derivação não cura
a hidrocefalia e o dano ao tecido cerebral permanece. Este procedimento não é
perfeito, pode ser mal-funcionante, pode coagular, causar infecção e mesmo se
quebrar.
Ventriculostomia -
Ventriculoscópios permitem que novas vias de líquor possam ser criadas no
cérebro, ou as antigas possam ser re-abertas.
quinta-feira, 8 de maio de 2014
Polimicrogiria
Polimicrogiria é uma
malformação da organização cortical que se caracteriza por múltiplos pequenos
giros separados por espessos e rasos sulcos. O objetivo do presente estudo
foi descrever as manifestações clínicas e eletroencefalográficas de pacientes
com polimicrogiria, que têm epilepsia e/ou distúrbio específico de linguagem e
dos familares dos pacientes com distúrbio específico de linguagem,
correlacionando-os com a neuroimagem. Os pacientes foram submetidos a exame
clínico e neurológico, com particular atenção aos sinais pseudobulbares, e
realizaram eletroencefalograma de rotina com até 4 horas de duração. Quando
possível, foram submetidos a vídeo-monitorização. Os dados de neuroimagem foram
classificados em: polimicrogiria perisylviana (subdividida em holosylviana,
parietal posterior bilateral, generalizada), polimicrogiria hemisférica e
polimicrogiria frontal (TEIXEIRA, 2006).
.jpg)

.jpg){kind=link}
Achados eletroencefalográficos:
Categorizados em:
- normal;
- anormal com atividade epileptiforme;
- anormal com atividade não
epileptiforme;
- anormal com atividade
epileptiforme e não epileptiforme;
- anormal com estado de mal
elétrico (EME);
- anormal com atividade
epileptiforme contínua e quanto à presença ou não de ativação da atividade
epileptiforme pelo sono.
Foram estudados 40 pacientes: 16 pacientes com
polimicrogiria holosylviana, 14 com polimicrogiria parietal posterior, quatro
com polimicrogiria generalizada, três com polimicrogiria hemisférica e três com
polimicrogiria frontal. Observou-se nos pacientes com polimicrogiria holosylviana:
sinais pseudobulbares em 11, hemiparesia em seis sendo um paciente com dupla
hemiparesia. Três possuíam deficiência mental e cinco tinham epilepsia. O
eletroencefalograma estava alterado em oito pacientes e atividade epileptiforme
foi registrada em seis, sendo que em dois foi registrada atividade
epileptiforme contínua focal. Entre os pacientes com polimicrogiria parietal
posterior bilateral, cinco apresentavam sinais pseudobulbares e um possuía
hemiparesia. Nenhum tinha alteração cognitiva ou epilepsia. Apenas um paciente
apresentou ao eletroencefalograma atividade epileptiforme focal. Todos os pacientes com polimicrogiria
generalizada tinham sinal pseudobulbar, deficiência mental e epilepsia. O
quadro motor estava presente em três pacientes e apenas um deles não
apresentava atividade epileptiforme no eletroencefalograma. Entre os pacientes
com polimicrogiria hemisférica, todos apresentavam sinal pseudobulbar e
hemiparesia, dois pacientes tinham deficiência mental e epilepsia, e um
paciente atraso do desenvolvimento neuropsicomotor. Nos registros
eletroencefalográficos, dois pacientes apresentaram EME focal. Os pacientes com
polimicrogiria frontal não possuíam sinal pseudobulbar e em um único paciente
foi detectada hemiparesia. Epilepsia estava presente em dois pacientes. No
registro eletroencefalográfico, dois exames apresentaram anormalidades
não-epileptiformes. Com os dados acima descritos foi possível observar que: os
sinais pseudobulbares foram mais freqüentes em pacientes com idade menor ou
igual a 15 anos; atividade epileptiforme e lentificação da atividade de base
teve associação com epilepsia e alteração cognitiva e o distúrbio específico de
linguagem apresentou uma relação inversa com estes achados
eletroencefalográficos; a maioria dos nossos pacientes apresentou exame
eletroencefalográfico normal; na polimicrogiria holosylviana, as atividades
epileptiformes predominaram na região fronto-temporal e as atividades não
epileptiformes predominaram na região fronto-centro-temporal; EME ocorreu em
polimicrogiria hemisférica e atividade epileptiforme contínua focal ocorreu em
polimicrogiria holosylviana bilateral assimétrica com provável displasia
cortical associada; a ativação da atividade epileptiforme pelo sono foi um
achado freqüente em nossa casuística; existe correlação direta entre as
manifestações clínicas e anormalidades eletroencefalográficas e extensão do
córtex polimicrogírico (TEIXEIRA, 2006).

Referências: TEIXEIRA, K.C.S. Clinical and electroencephalographic features of patients with polymicrogyria. Universidade Estadual de Campinas, Campinas -SP, 2006.
segunda-feira, 5 de maio de 2014
Fissura labiopalatal ( Lábio Leporino)
Lábio leporino é uma fissura congênita do lábio que pode ser
complicada por vezes com a divisão do processo alveolar, da abóboda palatina e
do véu do paladar.
As crianças com fissura labiopalatal (caso não associada a alguma síndrome) têm funções psicomotoras e desenvolvimento
normais, não devendo ser discriminadas.
O lábio leporino é uma anomalia que consiste
numa má formação do bebê que acontece durante a quarta até a décima semana de
gestação, ocasionando uma fissura entre o nariz e a boca. Essa má formação tem
diversos níveis e varia de uma pequena fenda até mesmo a separação total do
lábio superior no nariz até a boca, variando de leve a severo. Não
esqueça que os estudos comprovam que 50% das crianças que nascem com lábio
leporino também apresentam a fenda palatina que é quando o céu da boca também
não se fecha. Essas duas anomalias podem acontecer juntas ou separadas, e
afetam uma a cada 700 crianças, sendo mais frequentes entre crianças asiáticas
e índias americanas. Nas crianças afro-americanas a incidência de casos de
lábio leporino é significativamente menor. É importante mencionar que crianças
que nascem com lábio leporino ou fenda palatina não apresentam necessariamente
nenhum outro problema físico ou mental.

Causas da incidência do Lábio Leporino ou Fenda Palatina
As pesquisas médicas e cientificas acerca das causas que
ocasionariam essa má formação no feto foram infrutíferas, e ainda não se
conhecem as causas que justifiquem essa anomalia, entretanto há evidencias que
indicam que o problema acontece preferencialmente em famílias que tem em seu
histórico outros casos, seja no pai ou outro parente próximo, também a
possibilidade de se ter dois filhos com o mesmo problema também existe. Fatores
ambientais, tais como drogas, remédios, fungos, produtos químicos, deficiência
de ácido fólico e outras vitaminas podem interferir no fechamento
normal do palato e também no desenvolvimento dos lábios da criança.
 Detecção e correção
Detecção e correção
Hoje em dia, graças ao avanço da tecnologia e aos modernos
aparelhos de ultrassom já é possível diagnosticar o lábio leporino ainda antes
do nascimento, assim os pais podem ser preparados para receber a criança e
também orientados sobre o tratamento. Tão logo a criança complete 5 quilos, e
apresentando boas condições de saúde e beleza a primeira cirurgia
corretiva pode ser realizada. A cirurgia do palato no entanto só pode ser
realizada depois do primeiro ano da criança.


Amamentação e alimentação
Um dos maiores problemas enfrentados pela criança avantajadas que
nasce com lábio leporino é a dificuldade para se alimentar e também para mamar
no peito, assim a mãe deve ser orientada para persistir na tentativa de
amamentação, e sendo impossível deve tirar o leite e dar na mamadeira para
criança. O leite materno é de suma importância para que o bebe ganhe peso e
possa fazer a cirurgia com o mínimo de riscos possíveis. As dificuldades
naturais de alimentação causadas por esses problemas podem ocasionar nas
crianças problemas como otites, pneumonias, anemias e dificuldades no
desenvolvimento da linguagem e da fala.
Tanto o lábio leporino quanto a fenda palatina são anomalias
físicas que podem ser corrigidas através de cirurgias, mas precisam de atenção
dos pais e tratamento adequado, para que a criança possa ter uma vida normal, e
sem preconceitos.
Atuação dos profissionais Dentistas e Cirurgiões em Geral: O Lábio Leporino (ou fissura labial) pode ser restrita apenas ao lábio ou também envolver estrutura óssea (palato, céu da boca), então, o tratamento depende muito do grau de comprometimento, da idade e das partes envolvidas. O Cirurgião-Plastico normalmente cuida da plástica labial, e algums cirurgias de Exerto Ósseo e Ortognática (estruturas ósseas) quando necessário, são realizadas por Cirurgiões Buco Maxilo Faciais. Trabalham também, nas suas respectivas áreas, o Ortodontista, Odontopediatra, Otorrinolaringologista, Pediatra, Fonoaudióloga, dentre outros. Quanto mais complexo for o problema, normalmente mais profissionais estão envolvidos (equipe). O envolvimento dos profissionais depende do grau de extensão da Fissura.
Alguns cuidados ajudam a lidar com sua condição inicial:
- manter a criança em postura mais ereta durante a amamentação
evitando engasgar, oferecer refeições em menores quantidades e a intervalos
mais curtos, permitindo que elimine os gases deglutidos, acompanhamento
pediátrico efetivo com tratamentos e complementação alimentar sempre que
necessário.
A lábioplastia poderá ser realizada a partir do 3º mês de idade,
desde que a criança esteja com boa saúde, sem anemia (Hb =10 ou Hb > 10) e
peso mínimo adequado (igual ou maior que 3 Kg).
É uma cirurgia realizada sob anestesia geral de duração de
aproximadamente 2 horas e sendo necessário um período de internação de 24 a 48
horas, em geral.
Os cuidados no pós-operatório são de grande importância para
obtenção de melhores resultados. Entre eles:
* evitar que a criança chore muito principalmente nos primeiros dias de pós operatório.
* manter o lábio sempre limpo, livre de crosta de sangue, que
devem ser retiradas com o auxílio de água filtrada ou acrescida de água
oxigenada a 10 volumes.
* passar Ricofort pomada 2 vezes ao dia no lábio até a retirada
dos pontos que deverá ser feita por volta do 7º dia de pós-operatório
* no dia de retirar os pontos, a criança deverá estar de jejum de
5 horas (a retirada é feita sob analgesia/sedação)
* não usar bico ou chupeta / mamadeira nos primeiros 30 dias pós-
operatórios
* não deixar a criança levar as mãos ou dedo na boca, se necessário
usar aparelho de contenção (imobilização) nos braços, principal- mente até a
retirada dos pontos
* após a retirada dos pontos, a mãe deverá fazer massagens suaves
nas cicatrizes do lábio, durante 3 meses, para que as mesmas fiquem finas e
macias
* a criança não deverá tomar sol na face nos 3 meses após a
cirurgia (usar filtro solar, de proteção 30 ou maior, no lábio)
* usar medicação analgésica e as demais prescritas, rigorosamente
e caso a criança tenha algum problema alérgico, alerte ao seu cirurgião.
* deverá ser feito o controle médico, na criança, com 1 mês de
pós- operatório e por volta de 1 ano de idade, sendo que, em casos especiais,
este controle poderá ser em intervalos menores.
Referências:
http://www.agendar.no.comunidades.net/index.php?pagina=1158922494_11
http://drauziovarella.com.br/crianca-2/labio-leporinofenda-palatina/
Malformações Congênitas
O que são malformações congênitas?
Malformação é o nome usado para descrever os processos anômalos de formação e desenvolvimento de órgãos e tecidos do corpo humano. Congênito significa presente ao nascimento. Dessa maneira, malformações congênitas são alterações de desenvolvimento de órgãos e tecidos presentes desde o nascimento.
O que causam malformações congênitas?
As malformações congênitas podem ser decorrentes de uma infinidade de fatores causais. Esses fatores podem ser divididos em 2 grandes grupos: ambiental e genético. Muitas das alterações congênitas são decorrentes da interação conjunta tanto de fatores ambientais como genéticos. Apesar do embrião humano estar bem protegido no útero, certos agentes ambientais, chamados teratógenos, podem causar interrupções no desenvolvimento quando a mãe é exposta a eles. Teratógeno é qualquer agente capaz de produzir malformação congênita ou de aumentar a incidência de uma malformação em determinada população.
Quais são as causas ambientais de malformações congênitas?
A exposição do embrião ou feto em desenvolvimento a determinadas substâncias ou eventos durante a gestação podem causar malformação. Dentre os eventos durante a gestação, citam-se: uso de medicamentos (p. ex.: fenitoína, misoprostol, lítio) ou substâncias teratogênicas (p. ex.: álcool, drogas, andrógenos e progestógenos), infecções (p. ex.: citomegalovírus, varicela, HIV, Rubéola, Toxoplasmose), falta de nutrientes ou vitaminas (p. ex.: ácido fólico), dentre outras.
Quais são as causas genéticas de malformações congênitas?
Diversas alterações genéticas são relacionadas à gênese de malformações congênitas. Podem ser citadas: alterações dos cromossomos (p. ex.: síndrome de Down), alterações do número de cópia de segmentos cromossômicos (p. ex.: síndrome velocardiofacial), mutações deletérias em mais de uma centena de genes, dentre outras.
Quais são as malformações congênitas mais comuns?
As malformações mais comuns são: anomalias do sistema nervoso (p. ex.: espinha bífida, anencefalia, hidrocefalia), defeitos cardíacos (p. ex.: comunicação interatrial, comunicação interventricular) fendas oro-faciais (p. ex.: fenda laial, fenda palatina), alterações oculares (p. ex.: catarata congênita, glaucoma congênito, microftalmia), alterações das orelhas, alterações do trato digestivo (p. ex.: atresia esofágica, atresia anal), hérnia diafragmática, alterações do trato gênito-urinário (p. ex.: agenesia renal, valva de uretra posterior, hipospádia), defeitos dos membros (p. ex.: membros encurtados, pé torto congênito, polidactilia), alterações esqueléticas (p. ex.: displasias esqueléticas), dentre outras.
Como investigar as malformações congênitas?
O processo diagnóstico de malformações pode ser longo e demorado. Geralmente requer uma história detalhada do paciente e de sua família, interrogatório sobre o pré-natal, parto, período neonatal e evolução clínica. A avaliação também deve incluir um exame físico completo e minucioso e exames subsidiários com o intuito de se realizar uma busca ativa por malformações em outros órgãos ou sistemas. Após esta avaliação inicial, podem ser solicitados testes genéticos.
Como e quando são solicitados os testes genéticos?
Existem diversos tipos de testes genéticos, cada qual indicado para situações específicas. O teste genético deve ser solicitado a partir de uma hipótese diagnóstica clínica.
Referências
CLARSON M. Bruce. Tradução de Fernando Simão Vugman e Ithamar Vugman.
{kind=link}
Firth HV, et al. Oxford desk reference – clinical genetics. 2005. Oxford University Press, UK.
MOORE Keith L.. Embriologua Clínica. 5ª Edição. Guanabara Koogan 1996.
Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD).
Assinar:
Comentários (Atom)